A Human Cytome Project - discussion
![[construction icon]](https://www.vanosta.be/images/worker.gif)
Index
- A cytome project
- A proposal for the exploration of the human cytome - only high level concept
- A framework for the exploration of the human cytome
- Introduction
- Problems of drug discovery and development - problems at several levels
- Improving drug discovery and development - attempts at several levels
- Problems with disease models - models and the intrahuman ecosystem
- Less questions, but better answers - decomplexification of questions, improving answers
Vision
Attaining new insight in the biology of man, shaping medicine, biotechnology, drug discovery and development into a premier precision tool of the future, for the improvement of disease diagnosis, disease outcome prediction and curing the diseases of mankind.
Introduction
For no perfect discovery can be made upon a flat or a level; neither is it possible to discover the
more remote and deeper parts of any science if you stand but upon the level of the same science,
and ascend not to a higher science.
Francis Bacon, The Advancement of Learning (1605)
Basic and applied (pharmacological) science is an evolutionary process, driven by stochastics. Hypotheses are the mutations and the experiment is the environment in which they evolve. Hypotheses evolving on an island which does not represent the final biotope (man) in which they have to survive, mostly share the fate of the Dodo.
A human cytome project aims at creating a better understanding of a cellular level of biological complexity in order to allow us to close the gap between (our) molecules and the intrahuman ecosystem. Understanding the (heterogeneous) cellular level of biological organisation and complexity is (almost) within reach of present day science, which makes such a project ambitious but achievable. A human cytome project is about creating a solid translational science, not from bench to bedside, but from molecule to man. Even more than translational science, it is about transformational science as it transforms the subcellular realm into the intrahuman ecosystem. The human cell as a process is still out of reach for modern science.
Although the present model of pharmaceutical discovery and development is capable of coping with massive and late stage failures during the development life cycle of a new drug, it is not the way we should continue to work as the challenges ahead are even bigger than the ones we already faced in the past. The current process is capable to deliver enough drugs to sustain itself, given the disease mechanisms it deals with do not surpass a modest level of biological complexity. The scientific engine of the overall process is not yet capable to predictably deliver new drugs which can stand the challenges of biological complexity. At any given moment in time the actual performance of the scientific engine of the process and its scientists has to be taken more or less for granted, and only cost (labor, headcount, process engineering) and income (price, health insurance, government pricing policies) are available for marginal improvements in performance and productivity. We have to destroy too much capital and human effort to sustain the pipeline, which nowadays resembles a small tube in most companies. No matter how efficient we engineer our business processes, we cannot hide from the truth that we are running a process which has too much risk hidden underneath to feel comfortable with as the basis for delivering new treatments for the diseases of our society. Although the pharmaceutical process matrix is in itself consistently organised, it is is slightly out of touch with the complexity of clinical reality. This becomes visible at the end of the pipeline, when truth is finaly forced upon us and we can no longer hide from clinical reality. Changing the overall process however is a gargantuan endeavour as it is all about the core scientific engine in the first place, which is almost inert when it comes to paradigm changes.
This article is dedicated to the patients hoping and waiting for new treatments of unmet medical needs and
the improvement of existing diagnostic techniques and therapies. It is also dedicated to all the scientists
working in basic and applied research, working hard to deliver these new drugs and treatments. A lot of hard work
has been done, but a lot more is needed for the future. Improving medicine, drug discovery and development is not a
trivial endeavour and requires a multidisciplinary apporach and complex prosesses and projects.
Although this article is critical about the (evolution of the) overall drug discovery and
development process it also honours the individual contributions of scientists who have discovered and
developed drugs which save and improve the lives of many people. The purpose of critical discussion is to
advance the understanding of the field. While many are spurred to criticize from competitive instincts,
"a discussion which you win but which fails to help ... clarify ... should be regarded as a sheer
loss." (Popper). Let us look at the present with the future in mind.
(See also
Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products - USA,
New Safe Medicines Faster Project - Europe EU
and the
Priority Medicines for Europe and the World Project "A Public Health Approach to Innovation" - WHO).
[Note] The cell atlas initiative of Chan Zuckerberg Biohub (CZ Biohub) is now creating an atlas of the different cell types within the human body, which came as a pleasant surprise.
Personal note
My personal (former) interest in cytomics, grew out of my own work on High Content Screening, as you can see in:
- Image Analysis gives a clear view in research
- Scalespace or Differential Geometry
- Color Differential Geometry or the Spatial Color Model
- Application of linear scale space and the spatial color model in light microscopy
- Automated Tiled Multi-mode Image Acquisition and Processing Applied to Pharmaceutical Research
- The M5 framework for exploring the cytome
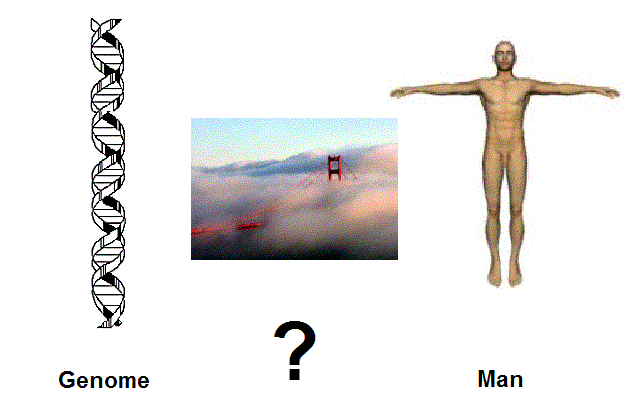
Figure 1. The bridge between the genome and the human biosystem is still not clear and the gap is still wide open. We desprately need to understand the dynamics of the molecules of man within their cellular environment, not just the ex-vivo molecules as such. But this remains a big challenge, which is at the frontier of present day science. |
The completion of the
Human Genome Project (HGP) holds
many promises for the understanding of the genetics of man
and the involvement of genes in human diseases. However the use of this
information has to be viewed from another perspective as is currently being
done, if we want to use this knowledge to improve medicine more efficiently. We have to build better
bridges from basic science to clinical applications and from molecules to man (Figure 1).
Predicting the dynamics of the cell and its fate in diseases from the genome
upwards is likely to fail due to the complexity of metabolic processing and
environmental influences on the cellular metabolism and the phenotype of the
entire organism. Reductionism cannot save us anymore, now we have reached the stage
at which we have to deal with the intertwined and multifaceted complexity of pathways from gene to organism.
Going "from genes to health" improvement requires an
understanding of disease processes beyond the boundaries of genomics and
proteomics. We need a better understanding of cellular physiology and beyond.
The "Book Of Life" is a novel, not a dictionary. Genes work in far
more complex ways than anticipated, understanding the relationship between
genome, cell biology and disease will take time and hard work.
In 2000, when he and his colleagues at Celera Genomics, in Rockville, Md.,
finished sequencing the human genome, J. Craig Venter announced the advent of the
"century of biology." I expect it to become the "century of applied biology" in order
to bring the treasures of the
Human Genome Project (HGP)
to clinical applications.
The complex clinical
reality of disease processes extends beyond the present-day disease models and the
(current) boundaries of basic and applied research. When we close the doors of our
labs behind us and as physicians are confronted with the clinical reality of
diseases in the outside world, our disease models fail all too often, as we can
witness in the diagnosis and treatment of complex diseases. This is also
painfully obvious in the dramatically high attrition rates during clinical
development of new drugs. A lot of work has been done, but even more is waiting ahead
of us. If we do not take up the challenge, the treasures of the
Human Genome Project
will remain hidden for too long.
Drug Discovery and Development
has to come up with drugs which can stand the
test of clinical reality in complex biological systems, but is being squeezed
between the failing (reductionistic) disease models and the demands for success of pharmaceutical
companies and society. Applied research has to provide the step stones to cross
the river from basic reductionistic disease models to complex clinical disease systems,
ideally without getting our feet wet or drowning before we reach the other side of the
river.
How do we close the gap
from model to clinic and find new directions for research?
The functional correlation between genome structure and clinically expressed
disease is too low to lead to functional predictions from the genome and even
proteome level upwards, without taking into account the spatial and temporal
dynamics of systems such as cells, organs and organisms. Pathological processes have to be
viewed from a higher organizational level of biology in order to capture the
dynamics of in-vivo processes involved in diseases.
The current bottom-up view
on genomic and proteomic research suffers from a correlation and prediction
deficit in relation to the entire system of an organism. The genome and proteome are the
omega of biological research, not the alpha of drug discovery or disease
treatment. From disease to gene we may find a link, but turning around and go
back to develop a treatment for the clinical disease fails in many cases. We
may find that a gene or genes may be part of a disease process, but we cannot explain
the entire disease process from the genome level alone. A gene may be involved
in a disease, but the entire disease process is not contained within the gene.
To discover the involvement of a gene or protein in a disease, does not predict
the potential for successful development of a treatment for the clinical
disease entity as such.
In-vivo variation is not an
artefact of life, but a fact of life. The extraction of the appropriate attributes of a biological process in
health and/or disease requires capturing the spatial and temporal dynamics of
its manifestations at multiple scales and dimensions of biological
organization. Disease entities express themselves in a space-time continuum in
which their physical and chemical attributes evolve in a highly dynamic way.
Capturing the appropriate features and disease describing parameters from the
background noise of their surrounding processes and structures is more
difficult than finding a needle in a haystack.
On
The idea of a Human Cytome
Project is already being discussed at scientific conferences, such as FOM 2004, ISLH
2004, ISAC XXII, EWGCCA 2004. At Focus
on Microscopy (FOM) in
Already several articles have been written on the
Human Cytome Project (Valet G, 2004; Valet G, 2004b; Valet G,
2004c; Valet G, 2005a; Valet G, 2005b) and
Cytomics.
As the idea of a Human Cytome Project
seems to have generated some interest in the scientific community, I decided to
put the original message and question on my personal website for reference, so
here it is.
Deliverables of a Human Cytome Project
The outcome of cytome research and a Human Cytome Project (HCP) should improve our
understanding of in-vivo patho-physiology in man.
It should:
- Reduce (late stage) attrition in clinical development
- Improve predictive power of preclinical-clinical transition
- Improve predictive power of preclinical disease models
- Improve predictive power of early diagnosis of disease
- Improve predictive power of disease models
We must achieve a better understanding of clinical disease processes in:
- Individual physiologicaly relevant celltypes
- The cytomes of model organisms
- The entire human cytome
What should we do to achieve this? Study pathological processes in:
- Individual cells whith physiological relevance
- Study the cytomes of model organisms and man in-vitro and in-vivo
- Build better correlation matrix between models and man
The rest of this article will discuss the key issues and the scientific reasons for a Human Cytome Project. The impact on pharmaceutical development is being discussed in drug discovery and development.
Overview of related articles on this website
Personal
interest and background where I provide som information how the idea for
a Human Cytome Project (HCP) has grown over time.
The original posting, on
Monday, 1 December 2003, of the idea can be found on this webpage.
References
have been put together on one page.
Overview of problems and questions
Scientific background about
the idea can be found this webpage
The potential impact on the
efficiency of drug discovery and development where I give an analysis
of the reasons for the unacceptable high attrition rates in drug development which
have now reached 9O%. Our preclinical disease models are failing, they look back
instead of forward towards the clinical disease process in man.
Overview of solutions and suggestions
A proposal of how to explore the human cytome where I give an overview of
the deliverables and the scientific methods which are (already) avalable.
A concept for a software framework
for exploring the human cytome is a high-level concept for large scale
exploration of space and time in cells and organisms.
Monday, 1 December 2003 10:57:46 +0100
Hi,
I was wondering if there is
already something going on to set up a sort of "Human Cytome Project" In
my opinion the hardware and most of the software seems to be available to set
up such a project? For the cellular level, light-microscopy based reader
technology would be very interesting to use?
Studying and mapping the
genome, transcriptome and proteome at the organizational level of the cell for
various cell types and organ models could provide us with a lot of information
of what actually goes on in organisms in the spatio-spectro-temporal space?
I have been thinking
(working) about a concept which could provide the basic framework
for exploring and managing this cellular level of biological organization
research on a large scale, but I would like to know if there is already some
thought/work going on in the direction of setting up an initiative such as a "Human Cytome Project" ?
This is just an idea, so I
am really interested to hear if there is something in it, or even if it is not
worth while what I just wrote.
Best regards,
Peter Van Osta.
Why a Human Cytome Project?
Human Genome Project
".. Nearly two centuries ago, in this room, on this floor, Thomas Jefferson and a trusted aide spread out a
magnificent map ... the map was the product of his courageous expedition across the American frontier, all the
way to the Pacific ... Today, the world is joining us here in the East Room to behold a map of even greater significance.
We are here to celebrate the completion of the first survey of the entire human genome. Without a doubt, this is the
most important, most wondrous map ever produced by humankind..."
President of the USA, Bill Clinton, June 20, 2000
The
Human Genome Project
(Lander ES, 2003; Venter JC, 2003) has set a new milestone in medicine and the
understanding of human biology (Guttmacher, A., 2002;
Guttmacher, A., 2003). Since its conception in 1986,
it has answered many questions, but it has also left us with more questions to
answer and it opened new horizons for exploration (Dulbecco R., 1986; Collins
F., 2003). The results of the Human Genome Project lead to a first estimate
that there are only about 34,000 genes in the human genome and by the end of
2003 the number was reduced to some 25,000 genes (Claverie
J.-M., 2001; Wright F. A., 2001; Pennisi E., 2003). Now at the end of 2004 the euchromatic sequence of the human genome is complete, the
number of genes is estimated to be about 20,000 to 25,000 (Collins FS, 2004).
The preeminent French scientist and 1965 Nobel laureate Jacques Monod, said in 1972
"Tout ce qui est vrai pour le Colibacille est vrai pour l'éléphant"
("What is true for
Escherichia coli
is also true of the
elephant").
At that moment this idea or hypothesis was deemed adequate to explain our observations
of the link between genotype and phenotype. The completion of the Human Genome project however,
has proven (once more) that this simplistic view of the genotype - phenotype relation is inadequate
to explain the complexity of this relation. This rather simplistic view on the genotype to phenotype relation
has proven to be less than successful in unraveling the complex dynamics of human diseases
(high late-stage attrition rates in drug development). We must always remain critical of the value of
an hypothesis regarding its adaquacy, internal coherence, external consistency and its fruitfulness.
As with all inductive reasoning, the long time needed to confirm or reject an hypothesis leaves us
vulnerable to much wasted effort in the mean time.
Induction or inductive reasoning, sometimes called inductive logic,
is the process of reasoning in which the premises of an argument support the conclusion, but do not ensure it.
It is used to ascribe properties or relations to types based on limited observations of particular tokens;
or to formulate laws based on limited observations of recurring phenomenal patterns.
The conditional acceptance of a hypothesis already
leads to a lot of activity because of the high risk to be left behind when the hypotheis proves to be true.
Before the completion of the Human Genome Project the focus on genes and the neglect of applied functional research
was a vaild option based on the state of science at the moment. It came as a shock that it would require
a lot of hard work to find out about the complex relation between genotype and phenotype.
The outcome of the
Human Genome Project has revealed that the processing of our genetic information is
much more complex than in Prokaryotes. As such the results of the
Human Genome Project, will have the same
impact on biology as
Einstein's
work had on the
Newtonean
world of physics. Our view on
biology has changed beyond what we had expected when the Human Genome Project started. The dynamics of
life are more complex, but also more fascinating than we could ever think of, before we had completed
the Human Genome Project.
The Caenorhabditis (C.
elegans) genome is comprised of over 18,000 genes. The fruit fly (D.
melanogaster) genome consists of about 13,000 genes and as such it has fewer
genes than C. elegans, although as an organism it is far more complex. Gene
number alone does not predict functional complexity. Although there is much
more variation in the sizes of the genomes, this is not reflected in the number
of genes.
The functional uncoupling
of the dynamics of cellular function to its genomic gene-count came as a shock.
The complexity and diversity of organisms is not reflected in the structural
complexity of their genomes alone, but to a large extent it is hidden in the
dynamics of gene expression and cellular processing. As there is no linear
relation between the complexity of an organism and the physical structure of
its genome, there is also no one-on-one relation between the phenotype of an
organism and its genome. Relatively small differences between organisms, such
as man and chimpanzee do result in large functional differences in gene
processing and functional expression.
The structural relatedness
of the human and chimpanzee genome, does not explain the large difference in
brain function for which gene expression profiles in the brain are a better
predictive instrument (Caceres M, 2003; Fortna A, 2004; Uddin M, 2004). Functional differences
between chimpanzee and man are more outspoken in the brain than in other
organs. Gene expression differences are more related to cerebral physiology and
function in humans than gene sequences. Epigenetic phenomena within individual
cells and differential processing in different cell types have more predictive
power than the piecemeal and one-dimensional gene sequence approach, when
applied on complex structures such as the brain (Wilson KE, 2004).
From single gene and
genome to the cell and beyond
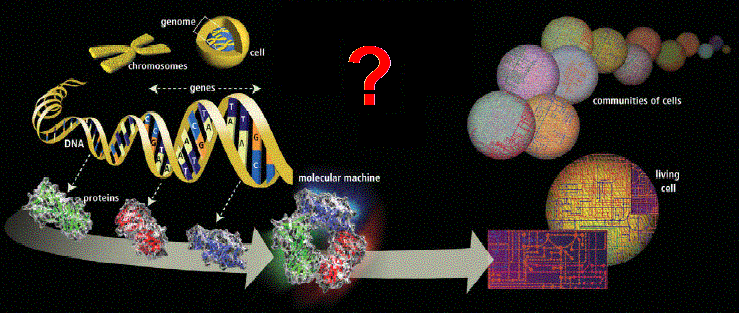
Figure 2. There is a lot of complex activity needed to build a complex cellular system (cytome) from its genes. Source: HGP media |
Now we are starting to use
the information coming out of the Human Genome Project, people start to
understand that the dynamics of the cell and its fate in disease processes
cannot simply be explained from its individual genes, genome or its proteome (Figure 2).
Although all cells in the human body share the same genome, there is
considerable heterogeneity in their phenotype and dynamics. Structural
information alone or information from too low an organizational level cannot
sufficiently predict higher-order phenomena as it does not sufficiently take
into account interactions at higher organizational levels and influences from
outside the low-level organizational unit. Cells have come up with compensation
mechanisms to maintain their structural and functional integrity in the face of
perturbations and uncertainty (Stelling J, 2004).
Organisms are capable of buffering genetic variation (Hartman JL 4th, 2001).
Genetic buffering mechanisms modify the genotype-phenotype relationship by
concealing the effects of genetic and environmental variation on phenotype
(Rutherford SL., 2000).
So if the structure of the
genome alone cannot explain the differences between species, disease processes
and the dynamics of the cell, where does our functional complexity and
interspecies differences come from? How do we continue in the post-genome era
to study the dynamics of the cell and entire organisms? How are genes related
to the function of an organism and where do we loose track? These questions are
not of academic importance alone, but their answers have a significant impact
on the diagnosis and treatment of (complex) diseases, drug discovery and
development.
Let us take a walk from
gene to protein and take a closer look at "The Central Dogma
of Molecular Biology", which I personally prefer to call an axiom instead
of a dogma. Science should only have axioms and leave dogmas to religion.
Associating genes with
diseases
In order to start studying
the contribution of a certain gene to a disease we must first find the gene(s)
which might play a role in a given disease. The strength of the association
must be detectable by the method being applied, which in complex gene-disease
relationships has to find the association on a background of significant
functional and phenotypical noise, such as in multifactorial diseases like
diabetes (Doria A., 2000). Variation in the
phenotypical expression of many quantitative traits (length, weight, ...) is due
to the simultaneous segregation of multiple quantitative trait loci (QTL) as
well as environmental influences. Genetic dissection of complex traits and
quantitative trait loci is a complex process (Darvasi
A., 1998; Darvasi A, 2002).A mono-factorial approach
is likely to fail in a multifactorial process of pathogenesis (Templeton AR.,
1998).
Giving a gene its place in
a disease process is not a trivial endeavour and it
is complicated by both technological and methodological difficulties.
Association studies offer a potentially powerful approach to identify genetic
variants that influence disease processes (Lohmueller KE, 2003; Roeder K,
2005). The density of Single Nucleotide Polymorphisms (SNP) makes them a
popular target for studying gene-disease associations. However it is not only
the density alone which counts, but also the information content of a given
polymorphism (Bader JS. 2001; Ohashi J, 2001; Byng
MC, 2003; Chapman JM, 2003; Garner C, 2003).
False positive correlations
of genetic markers with disease are reported due to a flawed statistical
analysis (Nurminen M., 1997; Edland SD, 2004; Wacholder S, 2004). In microarray experiments defining the
appropriate sample size to find differentially expressed genesis is an
important issue (Wang SJ, 2004). In complex diseases in which not only multiple
genes and the dynamics of gene products play a role, associating particular
genes with a disease entity is even more difficult than in so-called monogenic
diseases (Carey G., 1994; Long AD, 1999). Proper subgroup analyses in a randomised controlled trial (RCT) require careful design
(Brookes ST, 2001).
Turning a gene-disease association
into determining its role in the actual causation of a disease process is even
further away from finding and establishing a positive correlation (Templeton
AR., 1998).
From genome sequence to
gene activity
The genome sequence alone
does not allow us to predict the functional impact of sequence variations as
epigenetic modulation influences functional gene expression. Epigenetic
modulation of gene function is a cause of non-Mendelian inheritance patterns
and variability in the expression and penetrance of a disease. Even
transmission of an identical gene sequence is not a guarantee for identical
gene expression as the (in)-activation of a gene by epigenetic modulation
occurs differently when a gene is of paternal or maternal origin. Where (in what
cells or tissues) and when (at what stage of development or under what
conditions) genes are expressed is a highly dynamic process. The repression of
gene activity and the maintenance of the repressed state are fundamental
requirements of cell differentiation, ordered embryonic development and tissue
integrity (Czermin B, 2003). These spatial and
temporal gene expression patterns can be assembled into "localizome" maps (Dupuy D,
2004).
Epigenetic modulation of
gene expression is heritable during cell division but is not contained within
the DNA sequence itself (Reik W, 2001; Bjornsson HT, 2004; Kelly TL, 2004; Chong S, 2004). Epigenetic modulation is one of the
problems encountered when cloning, as the cloning process differs in its
epigenetic regulation of (embryonic) gene expression (Mann M, 2002).
This differential
inactivation of genes from maternal and paternal origin even leads to
functional X-chromosome mosaicism in women as their cells at random inactivate
one of their X chromosomes. X-inactivation occurs early in embryonic
development and all cells subsequent inherit a different functional X
chromosome. The inactivated X chromosome can be seen in a microscope as a Barr
body in the interphase nuclei of female mammals.
Differential activation of genes creates a functional chimera.
Chemical modification by
methylation of cytosine residues is a major regulator of mammalian genome
function and plays an important role in the intra-uterine development of an
organism and the regulation of gene expression (Urnov
FD, 2001). Tissue specific imprinting in genes leads to differential gene
expression in different tissues (Weinstein LS, 2001). Aberrant DNA methylation
has been implicated in the pathogenesis of a number of diseases associated with
aging, including cancer and cardiovascular and neurological diseases (Walter J,
2003; Jiang YH, 2004; Macaluso M, 2004). A dietary
component such as folic acid is a key component of DNA methylation during in
utero development, disease development and aging (McKay JA, 2004). Genes and
environment interact and this might play a critical role in the pathogenesis
and inheritance of complex diseases (Vercelli D,
2004).
Transcriptional regulation
in eukaryotes involves structurally and functionally distinct nuclear RNA
polymerases, corresponding general initiation factors, gene-specific
(DNA-binding) regulatory factors, and a variety of coregulatory
factors that act either through chromatin modifications or more directly to
facilitate formation and function of the preinitiation
complex (Roeder RG., 2005).
The gene expression flow
from mRNA to tRNA is not a smooth unregulated process in itself. Cells use
RNA-induced silencing complexes (RISCs) programmed
with small interfering RNA (siRNA) to knock down
target RNA levels (Wassenegger M, 1994; Robb GB,
2005). RNAi is used by Eukaryotes for sequence-specific, post-transcriptional
gene silencing (Cullen
The correlation of even a
gene sequence to the first steps in its expression does not show a one-on one
relation to the gene sequence itself. Modulators and regulators of
transcription and translation are showing a highly dynamic process regulation
mechanism. Cells use several mechanisms to create functional flexibility from
(relative) structural (genome sequence) rigidity. The genome is a repository of
our genetic potential, but only a part of it is active at different spatial and
temporal locations during our lifetime. It is not only important to know what
we can do within the limitations of our genomic boundaries, but also how we
deal with this potential in spatial and temporal patterns during our lives. We
do not deploy the full potential of our genome at every moment of our life and
in all our cells in the same way. Although all our cells share the same genome,
they are highly diverse in their structure and function, not only are they spatially
differentiated but also temporally. The relation of gene structure to its
function is a bidirectional process of which our understanding of the impact of
different modulators is still not sufficient to create highly correlating
disease models.
From gene to protein, a
bumpy road
A eukaryote, such as Homo
sapiens, has no one-on-one relation to its genes. The dynamics of gene
expression is regulated by hypo-, iso- and epigenetic operators. The gene may
be the structural unit of inheritance, but the protein domain is the functional
unit of metabolism.
When we talk about protein structure,
the primary structure refers to the amino acid sequence in a protein (1D). The primary
structure is most closely related to mRNA and as such the gene sequence and
gene structure from which the protein originates. The terms secondary and
tertiary structure refer to the 3D conformation of a protein chain. Secondary
structure refers to the interactions of the backbone chain (alpha helical, beta
sheet, etc.). Tertiary structure refers to interactions of the side chains.
Quaternary structure refers to the interaction between separate chains in a
multi-chain protein (4D). The combined shape of the secondary and tertiary
structure and the quaternary structure is referred to as the conformation of
the protein. With increasing dimensionality, the relation between a higher
order organization of protein structure and its gene relaxes as other physical
and chemical influences play an increasingly important role in its physical and
functional integrity.
In a mature enzyme, only a
relatively small number of its amino-acids interact with a ligand, the majority
of amino-acids help to create the appropriate 3D and even 4D structures
required for its in-vivo functionality. Structural proteins and enzymes may
show interactions over larger parts of their molecular surface to form
functional homo- or hetero-polymers in their quaternary structure. From a single
gene
to a protein, we have to deal with the dynamics of gene expression
regulation and mRNA formation (promoters, cis- and
trans-regulation, transcription, splicing). We have to deal with the
interaction of tRNA with mRNA in the translation of an mRNA sequence into a
protein sequence and post-processing of the protein sequence into a functional
3D and 4D structure (Wobble, sequence processing, protein folding and
interaction).
A structural similarity at
the genome level does not lead to functional similarity, due to epigenetic
regulation (Eckhardt F., 2004). Sequence variation, due to mutations does not
bleed through to the protein level one-on one. Basic mechanisms act as powerful
uncouplers of gene structure from protein function. Mutations in the DNA and
errors during transcription of the DNA-sequence into mRNA are not linear
predictive for the structure and function of the protein resulting from the
translation of the DNA-sequence into the protein-sequence, due to the
degeneration of the genetic code. The deleterious effects of sequence
variations are up to a certain extent suppressed by the Wobble-mechanism used
in base-pairing in translating mRNA to protein (Crick F, 1966).
Protein sequence = k x gene sequence
In this formula, "k" is
always smaller than one for most amino acids built into a protein, due to
mechanisms such as splicing variation, Wobble mechanism.
In eukaryotes, a relatively
simple genome compared to their functional and structural complexity can be
used, because of the existence of introns and exons.
An exon in general defines a functional domain and these domains are rearranged
to create a more complex proteome than the genome it is derived from.
Constitutive and alternative splicing of genes is dynamically regulated at the
moment of transcription and pre-mRNA splicing by cis-
and trans-acting factors (Kornblihtt AR, 2004; Sharp
PA, 1988). Before the completion of the Human Genome Project was finished it
was expected that man would need about 100,000 genes to explain the structural
and functional complexity of our species. This number has collapsed to about
25,000 genes and is about four times (75 percent) lower than expected (Collins
FS, 2004). The functional differences between species are more related to
differential processing, due to different up- and down regulation of genes in
different cell types and organs.
The use of different promoters
and splicing variants is used to tune protein and enzyme structure and function
in different cell locations and organs (Ayoubi TA, 1996, Masure
S, 1999; Nogues G, 2003, Yeo G, 2004). Promoter
variation and differential splicing allows for spatiotemporal differentiation
in protein expression, while the organism does not have to manage an explosion
in genomic size and sequence-complexity. This mechanism helps to uncouple the
protein from the rigidity of the gene sequence in order to allow for functional
variation while restricting structural variation at the genome level (Nadal-Ginard B, 1991). Functional differentiation in gene
expression allows for a better adaptability to changing conditions, without the
need for fast-paced changes in gene structure.
Protein folding of a linear
amino-acid sequence into a 3D protein also acts as a functional uncoupler of
gene sequence to protein function. Changes in the physical and chemical
environment of the protein may change the shape and alter the conformation of a
protein. By putting a protein in a different physical and chemical environment
which will change the ability of the van der Waals,
hydrogen, ionic and covalent bonds which hold the protein together in its
particular conformation, it is possible to cause the molecule to unfold by
breaking those bonds and make it change or even lose its function
(denaturation). 3D and 4D protein folding is a complex process. Even today the
protein folding problem remains one of the most basic unsolved problems in
computational biology. Predicting protein folding from the gene upwards ignores
the influence of the post-translational modification (PTM) and the influence of
the in-vivo physico-chemical environment of the protein. Proteoglycans and glycoproteins are not derived from a gene sequence as such,
but their structure is the result of extensive post-translational modification.
Cell membranes contain phospholipids, which are not encoded by DNA as such, but
they result from metabolic processing and nutritional components.
While the protein-sequence
at the moment of translation is related to the gene-sequence, the final
structure and function of an enzyme is in addition defined by
post-translational modification (PTM) and its physico-chemical environment
(Kukuruzinska MA, 1998; Uversky VN, 2003; Schramm A,
2003; Seddon AM, 2004). Studying protein folding is a
computational complex process and still the focus of intensive research (Murzin
A. G., 1995; Orengo, C.A., 1997; Dietmann S, 2001;
Day R, 2003; Harrison A, 2003; Pearl F, 2005). Epicellular regulation of
protein glycosylation also plays an important role in the dynamics of protein
activity (Medvedova L, 2004).
The majority of proteins
are subjected to a multitude of post-translational modifications.
Post-translational modification involves cleaving, attaching chemical groups
(prosthetic groups), internal cross-linking (disulfide bonds). Already more
than hundred different types of PTM are known, which act as functional
uncouplers of protein structure from the gene sequence (Hoogland C, 2004). A
protein precursor may be differently processed in different cell types and, in
addition, diseased cells may process a given precursor abnormally (Dockray GJ., 1987; Poly WJ., 1997; Rehfeld
JF., 1990; Rehfeld JF, 2003). Post-translational
protein modifications finely tune the cellular functions of each protein and
play an important role in cellular signaling, growth and transformation (Parekh RB, 1997; Seo J, 2004).
In a functional protein
only a very few specific residues are actually responsible for enzyme activity,
while the fold is much more closely related to ligand type (Martin AC, 1998).
The effect of an amino-acid change on protein structure and function depends on
the location of the amino-acid in the 3D structure, its physico-chemical
properties and the physico-chemical environment it is being processed and used.
Amino-acids which are distant neighbours in the
protein sequence can become close neighbours in the
3D structure of the protein and as such a protein sequence variation is only a
weak determinant of the function of a mature protein.
Proteins do not operate in
void, but they depend from other proteins and molecules for their function.
Proteins build complex cell signaling networks (CSNs)
in which the functional outcome cannot be predicted from each individual
protein alone (Berg EL, 2005; Eungdamrong NJ, 2004; Lengeler JW., 2000).
By just going from
DNA-sequence to 3D protein structure, the relation between genome sequence and
the functional status of a cell begins to fade. By taking this relation even
further from gene to organism, we lose additional predictive power.How will be able to design models that will
allow us to predict the functional outcome of a disease, when we use a fuzzy
model to start with? Powerful uncouplers of the structural relation of even a
protein to the gene it is primarily derived from, do not allow us to draw hard
conclusions about impact on the functional status of an organism from the gene
and genome sequence.
From proteome to cell
Eukaryotic cells
are highly compartmentalized; proteins do not exist in the cell as in a
homogeneous fluid, but in different compartments of the cell, each with a
different physico-chemical environment. The 3D and 4D structure of a protein
and its functionality is highly dependent from the in-vivo physico-chemical
environment of the protein. Cellular structure and metabolism is organized and
differentiated in both space and time.
Studying proteins without
taking into account their spatial and temporal organization in a cell, ignores
the complexity and dynamics of protein expression and interaction in a cell.
Studying proteins in-vivo reveals more about their function and dynamics (Chen,
X., 2002; Hesse J, 2002; Pimpl P, 2002; Viallet PM, 2003; Murphy R. F., 2004).
Without information about the relation between cellular structure and function,
a lot of information is lost. A 2D protein-profile may show the entire protein
content of a cell, but we lose all information about the intracellular spatial
and temporal distribution of these proteins.
Eukaryotic cells are highly
spatially differentiated structures. Proteins involved in trans-membrane
trafficking, require a membrane to do their work and cannot do their work
outside this specific physico-chemical environment. A protein has to reach the
appropriate physico-chemical environment in the cell in order to do its work
properly (Graham TR., 2004). Studying a protein outside its in-vivo
physico-chemical context leads to a loss of correlation with its in-vivo
dynamics.
There are three main
cellular compartments in a eukaryotic cell, the nucleus, cytoplasm and the cell
membrane. The nucleus itself is a highly organized 3D structure with highly
spatial and temporal differentiated DNA- and RNA-processing machinery (Lamond
AI, 2003; Politz, J., 2003; Pombo, A., 2003; Iborra
F, 2003; Spector DL., 2003; Cremer T, 2004). Both
transcription and splicing of the mRNA message are carried out in the nucleus (Sleeman JE., 2004). The distribution of eu-
and heterochromatin changes throughout the cell cycle, chromosomes and spindles
appear during cell division. The dynamics of gene transcription is visible in
the chromatin condensation patterns in the nucleus (Craig JM., 2005; Lippman Z, 2004). The nuclear envelope separates
transcription and DNA replication in the nucleus from the site of protein
synthesis in the cytoplasm (Rodriguez MS, 2004).
The cytoplasm itself
contains several organelles, smooth and rough endoplasmatic reticulum (SER and
RER), ribosomes, the Golgi apparatus, mitochondria, lysozomes and the cell
membrane. Each organelle deals with a different set of processes necessary for
cell development and maintenance. The membranes of organelles are highly
dynamic structures which undergo profound changes during the life cycle of a
cell (Ellenberg, J. 1997; Zaal, K. J. M., 1999). The
endoplasmic reticulum (ER) is a multifunctional signalling
organelle that controls a wide range of spatially and temporally differentiated
cellular processes (Berridge MJ., 2002).
The structural compartmentalisation of the intracellular environment
allows for a functional differentiation and provides a process flow management
mechanism. The membrane structure and the mitochondrial membrane potentials
(MMP) of mitochondria play an important role in their function. (Zhang H, 2001; Pham N.A, 2004). Microtubules play an
important role in cellular function and their organization and dynamics are
being studied by microscopy based techniques (De Mey J., 1981; De Brabander M.,
1986; Geuens G, 1986; De Brabander M, 1989; Geerts
H., 1991; Olson KR, 1999).
The dynamics of
intracellular ion-fluxes such as for calcium (Ca2+) is organized in
a highly dynamic and spatial and temporal complex pattern. Ions are themselves
not encoded by the genome, but play an important role in cellular function. The
intra- and extra-cellular dynamics of ions (concentration, flux) interact with
a spatial and temporally regulated pattern for protein expression and
differential protein activity. The complexity of intracellular
calcium-signaling extends beyond the mere expression profiles of genes encoding
the proteins involved in calcium-dynamics (Berridge MJ., 1981; Bootman MD,
2002; Cancela JM, 2002; Berridge MJ., 2003; Berridge
MJ, 2003b). For their proper function and survival cells have to manage Ca2+
concentration and flux in space, time and amplitude (Bootman MD, 2001). Calcium
is involved in the delicate process of spatially and temporally organization of
cellular communication (Berridge MJ., 2004).
As an example of spatial compartmentalisation in the cell, hydrolytic lysozomal
enzymes require a specific physical and chemical environment to do their work,
which inside the cell only exists inside the lysozomes (De Duve C, 1955). The
boundary membrane of the lysozome keeps the
hydrolytic enzymes away from the rest of the cytoplasm and so controls what
will be digested (De Duve C., 1966).
The cell membrane separates
the interior of the cell from its environment, but is a highly dynamic
structure (Kenworthy, A. K., 1998; Varma, R., 1998).
The appropriate spatial and temporal dynamics of the cell membrane are vital
for the survival of the cell. The cell membrane provides the physical
boundaries in which the cell can maintain a highly dynamic physical and
chemical environment. Cell-to-cell communication is dynamically managed at the
level of the cell membrane (Nohe A, 2004).
Proteins do their work in
spatially different cellular environments and with different spatial and
temporal patterns. A protein can be mobile in one cellular compartment and
immobile in another (Ellenberg J., 1997). Co-expressed proteins may in reality
never interact with each other because they do their work in separate cellular
compartments. The substrates of proteins may migrate through different cellular
compartments in order to be subjected to a highly dynamic interplay of
enzymatic processes. Proteins which do their work in the same cellular
compartment may only be expressed at different stages during the life cycle of
a cell. Spatial and temporal protein localization information can help us to
find entries into eukaryotic protein function (Kumar A, 2002).
An important temporal
differentiation of cellular processes occurs during the cell cycle. The
different stages in the cell
cycle each depend on the spatial and temporal expression of multiple
proteins. The passage of the cell through the cell cycle is controlled by
proteins in the cytoplasmic compartment, such as different Cyclins, Cyclin-dependent kinases (Cdks)
and the Anaphase-Promoting Complex (APC). First there is the G1
phase (growth and preparation of the chromosomes for replication). Secondly the
cell enters the S phase (synthesis of DNA and centrosomes) and finally the G2
phase which prepares the cell for the actual mitosis (M). The mitosis
itself consist of a spatial and temporal sequence of events, called the
prophase (mitotic spindle), prometaphase
(kinetochore), metaphase (metaphase plate), anaphase (breakdown of cohesins) and telophase
where a nuclear envelope reforms around each cluster of chromosomes and these
return to their more extended form.
However our understanding
of the cell cycle is still far from complete. The regulation of the cell cycle
by G1 cell cycle regulatory genes is more complex than we thought
(Pagano M, 2004).
Cells also operate in a
temporal pattern based on internal and external clocks. Cellular events must be
organized in the time dimension as well as in the space dimension for many
proteins to perform their cellular functions effectively (Okamura H., 2004).
Circadian molecular clocks regulate protein dynamics in temporal paterns (Crosthwaite SK., 2004; Hardin PE., 2004; Harms E,
2004; Hastings MH, 2004; Ikeda M, 2004; Rudic RD,
2004; Schwartz WJ, 2004; Shu Y, 2004; Takahashi JS.,
2004).In mammals there exists a central
circadian pacemaker which resides in the hypothalamic suprachiasmatic
nucleus (SCN), but circadian oscillators also exist in peripheral tissues (Yagita K, 2001).
We need to study and
understand the intracellular in-vivo dynamics of protein metabolism and its
spatial and temporal organization in different cell types. We need to study
intracellular protein ecology, not just ex-vivo protein interactions or
building a protein catalogue of only scalar dimensions. The spatial and
temporal patterns of intracellular protein dynamics are an important factor in
health and disease.
The dynamics of cellular
function
Taxonomy is the science of
organism classification and refers to either a hierarchical classification of
things, or the principles underlying the classification. Today the emphasis of
biological research is on classifying genes, proteins in large catalogues,
instead of studying the spatial and temporal dynamics of cellular processes in
vivo. The global analysis of cellular proteins or proteomics is now a key area
of research which is developing in the post-genome era (Chambers G, 2000;
Ideker T., 2001; Aitchison J.D, 2003). Proteins show functional grouping into
modules which can be grouped into elegant schemes (Hartwell, L.H., 1999; Segal,
E., 2003).
In-vivo however the spatial
and temporal distribution and interaction of proteins with other proteins,
substrates, etc., adds another layer of complexity which is not taken into
account by functional studies alone. Expression studies, no matter how we group
them, do not reveal the intracellular spatial and temporal distribution of
proteins and the functional outcome of their metabolic activity (spatial and
temporal substrate trafficking) in various cellular compartments. Studying
proteins only from a functional point of view ignores the impact of their
intracellular spatial and temporal dynamics.
The dynamics of cellular systems
can be explored in a global approach, which is now known as
systems biology.
Systems biology is not the biology of systems, it is the region between the
individual components and the system. It deals with those emerging properties
that arise when you go from the molecule to the system.
Systems biology is the in-between
between physiology or holism, which study the entire system, and molecular biology,
which only studies the molecules (reductionist approach). As such systems biology is
the glue between the genome and proteome on one side and the cytome and physiome
on the other side. The top-down approach of cytome and physiome research and the bottom-up
approach of genome and proteome research meet each other in systems biology.
I took me a while to come to terms
with systems biology, as I was trained (eighties of the 20th century) in medicine and
molecular biology in a traditional way. Systems biology studies
biological systems systematically and extensively and in the end tries to
formulate mathematical models that describe the structure of the system (Ideker
T., 2001; Klapa MI, 2003; Rives A.W, 2003). The end-point of present day systems biology
only takes into account infra-cellular dynamics and leaves iso- and epi-cellular
phenomena to "physiology". A "systems", but top-down, approach to
cytomics and physiomics is feasible with the technologies which are now emerging
(e.g. HCS, HCA, molecular imaging,..).
Studying the physics and chemistry of protein interactions cannot ignore the spatial and temporal
dynamics of cellular processes. We study nature "horizontaly", e.g. the genome
or proteome, while the flux in nature goes "verticaly", through a web of
intertwined pathways evolving in space and time. The focus of traditional -omics research
(genomics, proteomics) is perpendicular to the flow of events in nature.
The resultant vector which signifies our understanding of nature is aligned with the
way we work, not with the true flow of events in nature.
Molecular taxonomy or systems biology (genomics, proteomics) will not provide us
with all the answers we need to know, it is however an important stepstone
from molecule to man.
The cell is at the
crossroads of life itself, being the lowest order functional unit operating in
a functional complete way. It is the basic object of nature. As such the cell is for life what the atom is for
physics, the smallest biological level of organization, operating as a
functional unit. The cell doctrine
states that cells form the fundamental structural and functional
units of all living organisms and was proposed in 1838 by Matthias Schleiden and by Theodor Schwann.
Dysfunctional cells by whatever cause, either gene and/or protein
malfunction, infection, nutritional or environmental problems will eventually
cause the entire organism to lose its functional integrity. The dynamics of
cellular systems allow for the adaptation of the cell to a wide variety of
conditions and challenges, a relatively uniform physical structure combined
with a web of interacting dynamic processes leads to the multitude of cells
which we see in living organisms. In a living organism there is no such thing
as an average cell type from a functional point of view. Cells are functionally
highly diverse in both spatial and temporal dimensions.
The stochastic variation of
cellular processing at the molecular level is another cause of functional
uncoupling of the cytome from the genome and ads to the variability in
functional behavior between cells (McAdams H.H., 1999; Raser
J.M., 2004). Structural research alone underestimates the complexity of dynamic
processes as it does not capture sufficiently the dynamic complexity of the
cell. The dynamic interaction of processes in multiple pathways is the
centerpiece of cellular life, not the individual components or even individual
enzymatic reactions in the cell. There is no monotonic sequence of causation
from genome structure to cellular dynamics.
Cellular function can be
compared to a symphony in which multiple "instruments" contribute to a complex,
but in a healthy state harmonic, "sound".
Genes and the dynamics of
disease processes
The challenges faced by the
medical world today are no less today than the ones we faced a century ago. The
spectrum of diseases may have changed through time, as degenerative diseases
and cancer play an increasing role in modern society. On the other side an old
enemy is back on the rise, how much we thought that infectious diseases were a
thing of the past; they are back and with a new and frightening face.
Our increase in the
knowledge of the involvement of our genes and large scale proteomics in disease
processes has not lead to an increase in the productivity of pharmaceutical
research (Drews J., 2000; Huber,
In the case of diseases
where we have already found a genetic basis, this does not always allow us to
create a model for the disease process. To discover the involvement of a gene
in a disease process does not tell us anything about its place and relative
importance in the multiple and multilevel elements involved in the causation of
a disease, such as genes, nutrition, infectious agents and the environment. To
discover a causative element is not the same as understanding and predicting
its dynamic involvement in a disease process. What we do know is that all
causation has to pass through cells, as they constitute the "quanta" of the
organism itself.
Many diseases of clinical
importance have heterogeneous mechanisms which lead to the disease and only in
a subpopulation the diseases can be traced back to a single gene. In most cases
a multiplicity of mechanisms contributes to the diseases process. Genetic
information has a high predictive value in only a minority of cases.
Non-coding sequences,
inter-gene and epigenetic interactions have a significant impact on the
prediction of the age of occurrence, severity, and long-term prognosis of
diseases (El-Osta A., 2004, Perkins DO, 2004).
The importance of the
dynamics of the cell and its involvement in pathological processes and current
therapeutic efforts also requires a better understanding of its function and
phenotype in its relation to pathological processes in diseases, such as in cancer, Alzheimer
disease and infectious diseases,
such as AIDS, tuberculosis (TBC), influenza (flu), etc.
Trying to predict a disease
process from the genome (proteome) upwards, is like trying to solve a higher
order polynomial while omitting the majority of elements and expecting that the
equation will work:
e.g.: Disease process = a x x + b
Instead of using a higher
order multi-dimensional model, closer to in-vivo functional dynamics in which a
matrix or web of causation and consequences interacts in a high-dimensional
space-time continuum:
e.g.: Disease process = a x un
+ b x vo
+ c x wp
+ d x yq
+ e x zr
In addition, each parameter
which is being used in an equation is in itself the result of an underlying or
"overlying" dynamic process. Each layer of organization can be fed into higher or
lower order levels of organization as there is always a cross-influence in both
directions. It is a matter of expanding or collapsing the set of parameters and
taking into account or ignoring underlying "modifying" influences. Reducing the
complexity allows for a better understanding of a simplified model, but has a
decreased match to the complexity and dynamics of biological reality. When we
create a model, we should not regard it as a one-on-one substitute for reality
which we capture only partially into our model.
Infectious diseases
Infectious diseases still
pose a significant threat to the health and well being of (modern) society.
After years of relative neglect, nations are increasingly aware of the present
and future threats of infectious diseases and are even setting up new agencies,
such as the European
Centre for Disease Prevention and Control (ECDC) or expand the role of
existing organizations, such as the Centers for
Disease Control and Prevention (CDC). Beside their political and economical
impact on society, how do we deal with infectious diseases in science?
In infectious diseases the
environment, in this case the infectious agents, interacts in a complex way
with the host defense system of which much remains to be explored. We must be
aware of the fact that the golden era of antibiotics is already behind us as
many infectious agents (e.g. TBC, MRSA and other bacterial diseases) are
showing an increasing resistance against most classes of antibiotics which are
available today (Davies J, 1994). We have succeeded in less than a century to
destroy our best weapons against infectious diseases, due to misuse of
antibiotics both by physicians and their patients. Only the elderly remember
the days when mortality due to infections was a major cause of premature death,
but the moment is approaching when this nightmare will return. Emerging
infectious diseases (EIDs) and re-emerging infectious
diseases challenge our defenses (Ranga S, 1997; Fauci AS., 2004; Morens DM,
2004).
Viral diseases (e.g. AIDS,
influenza) are even harder to fight as they use the cellular machinery of the
body itself to reproduce. We need to study the pathological process in cells in
more detail and in a different way, in order to have a chance to succeed in the
new therapeutic challenges ahead of us. Viruses, under selective pressure of
modern antiviral drugs are also showing increasing resistance to treatment. We
are running out of time in our battle against infectious diseases and a
systematic approach will only give us the answers when it will be too late. We
are not setting the agenda, but the diseases are taking the lead.
Due to modern technology,
the time to respond to a new infectious challenge is being reduced. In modern
times, diseases take planes too, which makes it even harder to fight them by
classical isolation or quarantine. Airplanes may be safe to travel with,
compared to other transport systems, but they can cause secondary mortality by
transporting pathogens over large distances at a speed unknown to previous
generations, which gives a new meaning to airborne infections (Gerard E, 2002;
Van Herck K, 2004; Blair JE, 2004). Infectious
diseases may initially go unnoticed in underdeveloped areas of the world (e.g. Ebola virus Lassa fever,
Marburg virus), but as soon as they board a plane, it is modern technology
which will give them free access to the world (Clayton AJ, 1979; Gillen PB,
1999). A relatively long incubation time combined with a high mortality rate
will allow a disease to spread widely and cause a pandemic, before we even can
start a treatment program. If an unknown disease causes such a pandemic, we may
run out of time before we can find a cure as we first have to develop a
diagnostic tool. A recent example which is a model of what can happen was the
Severe Acute Respiratory Syndrome or SARS
(Peiris, J.S.M. 2003, Berger A, 2004; Heymann DL,
2004; Tambyah PA, 2004).
Robert Koch presented his
work on Tuberculosis on 24 March 1882 before the members of the Berlin
Physiological Society, which meant a breakthrough in the understanding of this
terrible disease (Winkle S, 1997, pp. 137-141). Now after more than 100 years
of research and drug development, TB is on the rise again. In the war against
infections such as Tuberculosis, there are no easy wins. We may win a fight but
for the majority of pathogens we can only reach a status quo, but never
completely win the war. Variability by mutating is a powerful weapon against
our drug treatments and pathogens use it to their great advantage.
We must keep our defenses
up to date and changing in order to outsmart our bacterial and viral enemies.
New antibiotics are not found within the human genome. Penicillin was
discovered by accident and many important antibiotics were found at the most
unlikely places (Fleming, A, 1929). No hypothesis or model can be formulated to
find the unexpected, but we have to find new antibiotics as bacteria are
closing in on us and some of our worst enemies are even winning the race.
Scientists are waiting with
fear for the next influenza pandemic which will hit us some day (Gust ID, 2001;
Capua I, 2004). Scientists are trying to understand
the lethal potential of the deadliest influenza epidemic of all times, which
occurred after the first World-War. Soon the virus which caused the influenza
pandemic, called the "Spanish flu" will re-emerge out of the test tubes of the
laboratory. Recent outbreaks of avian
flu have given us a preview of what can happen and evidence is increasing
that the possibilities for spreading avian influenza A virus (H5 or H7 subtype)
are worse than previously was assumed (Koopmans M, 2004; Kuiken
T, 2004).
New pathogens can have a
devastating effect on a human population. Examples of what can happen when a
new infectious agent hits a population with little or no immunological
"experience" with a (re-)introduced pathogen, can be found in the histories of
indigenous people confronted with infectious diseases introduced by European
colonization as in Australia
and Tasmania. Within 100 years of European colonization the total
population of full-blood Aboriginal people in Tasmania became extinct.
Introduced infectious diseases killed many more Aborigines than did direct
conflict. Infectious diseases such as smallpox, measles, and influenza were
major killers and even chickenpox was deadly as the Aboriginals had no
immunological history even with chickenpox. Of the 90 percent of the Aboriginal
population that died out as a result of European contact, it is estimated that
around 80 or 90 percent of the deaths were the result of disease.
Most people have no idea of
the role smallpox played in the destruction of an entire civilization after it
was brought to America by the conquistadores. About 50 to 90 percent of the
Native American population died of smallpox and the speed at which people died
is beyond our imagination (McMichael AJ, 2004; Winkle S., 1997, pp. 855-861). A
mortality of 50 percent for a new disease, for which we have no immunity, could
kill half of the population of a country or an entire continent. Western
society now has to fear the introduction of new pathogens from distant places
and when the disease has the right pathological profile; it will spread extensively
into the population before it is being diagnosed (e.g. AIDS). Re-emerging
infectious diseases are a global problem with a local impact. It is an
unpleasant thought that this time we will face the fate of the indigenous
people during European colonization.. In
modern times we not only have to fear the accidental spreading of infectious
diseases, but bio-terrorism will challenge our defenses sooner or later
(Broussard LA, 2001, Gottschalk R, 2004).
Finding the infectious
agent for a new and unknown disease requires something else than sequencing a
genome as this approach only works when we have the time to do the sequencing
while the pathogen takes its course. Analyzing the genome sequence of a new
infectious agent can only start after it has been isolated by more traditional
means (Berger A, 2004). Once we know the new pathogen, we can use its genome
sequence to develop rapid diagnostic tools, based on PCR, but in order to do
this we must first isolate it from the patient. Developing a therapy after this,
takes much longer and the genome sequence itself without additional functional
information is not enough. Only after Koch's postulates had been fulfilled, the
WHO officially declared on 16 April 2003 that a previously unknown coronavirus was the cause of SARS.
Modifying the disease
progression requires an interaction with the actual disease process which
extends beyond understanding the genome structure of the pathogen. Focusing
more on the dynamics of the interaction of cellular systems with pathogens and
using tools for functional research of the disease process at the cellular
level (and beyond) will hopefully allow us to respond in time when we are faced
with an unknown pathogen.
When we do not already have
an antibiotic, antiviral drug or vaccine at hand at the moment a new disease
hits us, either by accident or on purpose in biological warfare or bioterrorism, we are in serious (and lethal) trouble. In
this case the only thing left is the medieval solution of quarantaining
the infected people, which only works if we are able to contain them before
they spread over a country or even the planet (e.g. Ebola, SARS or HIV).
Although all cells in the
human body may share the same genome, there is a high spatial and temporal
differentiation in gene expression and metabolic dynamics in different cell
types and organs. In HIV, it is the CD4 lymphocytes which express the receptors
by which the virus can enter the cell (Fauci AS, 1996). A hepatocyte
may share its entire genome with a CD4 lymphocyte, but it does not express the
proteins encoded by the gene which allows the virus to enter the cell. The
progress of a HIV infection is also a highly dynamic process of interaction
between the host and the virus (Wei, X., 1995). The observation of differences
in disease progress leads to the discovery of a genetic restriction of HIV-1
infection and progression to AIDS by a deletion allele of the CCR5 structural
gene (Dean M, 1996). The emerging picture on infectious diseases is one of
highly polygenic patterns, with occasional major genes, along with significant
inter-population heterogeneity (Frodsham AJ, 2004). The complex interactions
and regulation of the Interleukin-1 (IL-1) family of proteins is just one of
the issues in elucidating the dynamics of the human immune system (Laurincova
B., 2000). Innate immunity represents the first line of defense against
invading pathogens and noxious stimuli.
Clinical observations lead
to genetic conclusions, but the way back to clinical treatment of diseases is a
long and winding road for which the gene sequence or protein structure does not
provide us with all the necessary information about the dynamics of the disease
process. Studying the cellular dynamics of disease processes provides us with
one of the step stones from gene to clinic. By focusing on genomics and
proteomics alone, there remains a correlation and predictive deficit in our
disease models.
Mendelian diseases
Mendelian inherited and
monogenic diseases have always been at the center of attention in the relation
of genetic variation to diseases. Monogenic diseases served as a model to prove
the use of genetic information to the development of a disease and the outcome
of a disease process. Phenotype-genotype relationships are complex even in the
case of many monogenic diseases. Increasingly complex interactions have now
been demonstrated in a number of monogenic Mendelian diseases (Nabholz CE, 2004). The (phenotypical and functional)
expression and development of even a monogenic disease depends on its context,
which comprises both other genes and environmental factors. These inter-gene
and epigenetic interactions have a significant impact on the prediction of the
age of occurrence, severity, and long-term prognosis of even "genetic" diseases
(Cajiao I, 2004; Hull J, 1998; Frank RE, 2004; Salvatore F, 2002; Sontag MK,
2004; Sangiuolo F, 2004). Understanding the root cause of disease does not
necessarily translate into developing a successful drug. The fact that there
are still no cures for classic genetic diseases such as muscular dystrophy,
cystic fibrosis, and Huntington's disease, the genes for which were
discovered 10 to 15 years ago, does not bode well for more complex diseases,
where the respective roles of genes and environment are harder to dissect.
The beta-thalassemias show
a remarkable phenotypic diversity caused by the action of many secondary and
tertiary modifiers, and a wide range of environmental factors (Weatherall DJ.,
2001). Sickle cell anaemia and cystic fibrosis can
serve as an example that genotype at a single locus rarely completely predicts
phenotype (Summers KM., 1996). Although the gene defect in Huntington's disease
is known for years, the contribution of the gene defect to the functional out
come of the disease is not yet known (Georgiou-Karistianis
N, 2003). Cell based research will help to elucidate the disease mechanism in
Huntington's disease (Arrasate M, 2004).
In cystic fibrosis, the
severity of the disease cannot be linked one-on-one to genetic variation in
CFTR (Grody W, 2003). Cystic fibrosis is the most
common autosomal recessive disorder in Caucasians, with a frequency of
approximately 1 in 3000 live births, so finding a cure for this disease has a
high impact on our society. Success stories with rare diseases may sound impressive
from a scientific point of view, but there is no escape from the economic
reality of the size of the patient population. So let us take a closer look at
cystic fibrosis as it is a disease of which the gene held responsible for the
disease was identified about 14 years ago (Rommens
JM, 1989; Collins FS., 1990).. The method
(reverse genetics) used to identify the gene, did not require an understanding
of the gene function at that moment or any understanding of the impact of
genetic heterogeneity on the phenotypical expression of the disease (Iannuzzi MC, 1990; Audrezet MP, 2004). By starting form the
gene for a single genetic disease such as cystic fibrosis, where did we get
after 14 years of hard labour?
A once "monogenic" disease
such as cystic fibrosis shows remarkable phenotypic variation and clinical
variation (Decaestecker K, 2004). By now about 1000 gene mutations of the
cystic fibrosis transmembrane conductance regulator
gene (CFTR) have been identified, which leads to a highly variable phenotypic
and clinical presentation of the disease. (McKone EF, 2003). Mutations in the
CFTR gene have been classified into 5 functional categories (Welsh MJ, 1993). A
list of 1000 mutations is reduced to 5 functional classes at the protein level,
which leads to a ratio of 0.5 percent for each mutation to lead to a distinct
CFTR chloride channel dysfunction. Due to the functional uncoupling of gene
structure to protein function in cystic fibrosis, genetic sequence variation
has a low impact on functional variation on the protein level (1000 to 5). More
important than gene sequence variation is the spatial location of a mutation in
the 3D structure of a protein. (Rich DP, 1993). Even
more important is the cellular and organ location of a functional defect as in
Cystic Fibrosis mainly the pathological process (Pseudomonas aeruginosa
infection) in the lungs are a major cause of morbidity and mortality (Elkin S,
2003).
Other genes act as
modulators of the disease outcome, even in a disease such as cystic fibrosis,
once regarded as a monogenic disease (Hull J, 1998, Frank RE, 2004; Salvatore
F, 2002; Sontag MK, 2004; Sangiuolo F., 2004). We even need to take into
account epigenetic information and environmental influences on disease outcome,
even in a so called monogenic disease as cystic fibrosis.
Human populations show
considerable genetic heterogeneity (allelic variation) and even geographic
variation, which leads to difficulties in using gene sequence based diagnostic
tools (Liu W, 2004; Raskin S, 2003). So, the sequence
of one individual's genome allows studying one person's genetic profile, but
does not lead to a population-wide prediction of genetic profiles. Genetic
heterogeneity uncouples clinical outcome from model gene sequences (Imahara SD,
2004). This problem is not solved by simply adding more sequence information
without a functional understanding of the meaning of sequence variation on
phenotypic expression and disease outcome in the patient. Structural
information without functional understanding leads to predictive deficits. The
functional understanding of a disease process must be at the level of the
patient and his cells and not at a lower order organizational level, such as
the genome or proteome alone.
Genetic heterogeneity leads
to a reduced sensitivity and an increase in false negative results if a genetic
test is not adapted to this genetic heterogeneity. A mutational test leads to a
simpler almost "binary" readout, instead of the more "analog" interpretation of
a continuum of values in a functional test, but this comes at a price. A test
which detects a disease marker at a higher organizational level can detect a
disease more easily and will lead to less false negatives in this case.
The complexity of even
monogenic diseases and the web of functional interactions between at the genome
level, protein interactions and environmental influences on the disease outcome
will dilute the predictive power of structural sequence information and the
DNA-level. Using low-dimensional intracellular data to predict iso- and
epicellular phenomena has a low predictive power to be used in clinical
situations as such.
No pharmaceutical company
would like the idea that it requires 14 years of preclinical research to reach
an IND after a new drug target was identified as in cystic fibrosis. Even if
only 1000 genes out of our 25,000 were involved in human diseases and would
require the same amount of work, it would take us the equivalent of 14,000
years of work on the scale as was needed to achieve the same results as for the
cystic fibrosis gene. But up to this moment no causal (gene) therapy came out
of the identification of the CFTR gene, but an improvement of prenatal
diagnostics (Klink D, 2004).
Pseudomonas aeruginosa lung
infection is the major cause of morbidity and mortality in patients with cystic
fibrosis (Elkin S, 2003). Over the past decades we have seen an improvement of
symptomatic therapy, but still no causal therapy, leaving aside a lung
transplant.
How are we going to develop
drugs which have a large enough patient population to pay for the costs of drug
discovery and development if we need to target individual mutant protein
molecules? If it can be so difficult to go from a single gene to develop a
therapy based on genetic information, how do we expect to proceed for the
entire genome and proteome?
Degenerative diseases and cancer
The increasing longevity of
western population is increasingly straining public healthcare systems, due to
an increase in incidence of degenerative diseases and cancer. A diminishing active
population has to support the growing financial demands of a healthcare system.
Improving the health and self-reliance of the growing number of elderly people
by efficient treatments of degenerative diseases and cancer is an important
political issue. Where are we and where are we going to in science to solve
these fundamental problems of modern society?
Unraveling the pathological
mechanism of a complex disease is a major scientific challenge and still beyond
reach of present day science in many cases. For degenerative diseases, such as Alzheimer disease , cancer, birth defects, cardiovascular
diseases, Parkinson's disease, diabetes, and nerve degeneration it is the
dynamics of the cellular machinery itself which fails. Sharing one genome does
not lead to sharing the same pathology, as cellular differentiation leads to a
highly diverse spatial and temporal cellular function and morphology.
Differential and heterogeneous degeneration patterns of different cell types
are the consequence of a highly differentiated spatial and temporal expression
pattern of proteins in different cell types and different sub-cellular
compartments.
Unravelling part of the genetics of a disease
does not yet bring therapeutic success. Multiple genes and (multiple)
environmental factors contribute to the disease process and its clinical
outcome in complex diseases (Liebman MN, 2002). In
Crohn's disease the gene defect found does not explain the severity of the
disease (Peltekova VD, 2004).. In breast cancer genetic variants of BRCA1
and BRCA2 do not have a consistent level of penetration and as such their
presence alone does not explain the disease process (Ford D et al, 1998;
Hartge, 2003). Although there is evidence for the involvement of the gene for
PPAR-gamma in type 2 diabetes is, the mechanism by which it contributes to the
disease process of diabetes is not clear and could not be deduced from genetic
information alone (Barroso I, 1999).
In APC (Adenomatous
Polyposis Coli) and HNPCC (Hereditary Non-Polyposis Colorectal Cancer) a
genetic origin, only accounts for about 5 percent of all cases of colorectal
cancer (Kinzler, 1996). Genes which are involved in diabetes, such as GCK
(glukokinase) , HNF1A and HNF4A (Hepatic Nuclear
Factor) are linked to less than 5 percent of cases of diabetes (Edlund, 1998, Fajans, 2001).
On of the major emerging
health problems of modern society is Alzheimer's disease (AD). This is not only
because widely known people, such as the former president of the USA, Ronald
Reagan, suffered from the disease in a long and unpleasant disease process.
Today AD is still a chronic disease without a cure which causes patients to
receive long-term care (Souder E, 2004).
Presently available drugs
improve symptoms, but do not have a profound disease-modifying effect and fail
to alter the course of AD, so it may be time to change the way we think about
AD therapeutics (Crentsil V., 2004; Citron M., 2004; Kostrzewa
RM, 2004)? Will we see a breakthrough in the understanding of the cellular and
molecular alterations that are responsible for the degeneration of neurons in
AD patients (Mattson MP., 2004)?
In Alzheimer's disease
(AD), only a minority of cases can be linked to a single hereditary gene
mutation, the complexity of the disease process extends beyond our present
understanding and disease models (Selkoe DJ., 2001; Eikelenboom
P, 2004). Neurodegeneration in AD may be caused by deposition of amyloid beta-peptide in plaques in brain tissue (Amyloid Hypothesis), but no causal treatment has come out
of this in 10 years of hard work (Hardy J, 2002; Lee HG, 2004; Lee HG, 2004b).
Little is understood about the dynamics of. amyloid
beta-peptide and its fundamental role in the disease process of AD (Regland B,
1992; Koo EH., 2002; LeVine
H 3rd., 2004).
A complex disease requires
studying and understanding a complex in-vivo pattern of a spatially and
temporally changing metabolic process, which goes beyond studying gene
expression profiles, either single or multiplexed. Studying the multi-scale
spatial and temporal dynamics of a complex disease process in a long-term
space-time continuum is a tremendous scientific challenge. Instead of focusing
on individual (molecular) targets in drug research and therapy, complex
diseases may require pathway-engineering or cell replacement to restore the
appropriate dynamics of spatial and temporal patterns of intracellular
molecular processes. Functional or structural protein (re-) modeling or
restoration in-vivo may be a better approach for complex diseases than just
docking a small molecule to an active binding site?
At this moment the cell is
the target for many therapeutic efforts to come to a causal therapy of complex
diseases, which we can now only treat with external substitution, such as diabetes. Many diseases are far more
complex and multi-factorial than monogenic diseases and should be studied with
more power at a higher biological level than the genome or proteome to capture
the complexity of the disease process.
One of the most promising
domains of research today is stem cell
research (He Q, 2003; Doss MX, 2004). Since the isolation and growth in culture
of proliferative cells derived from mouse embryos in 1981, stem cell research
has come a long way (Evans MJ, 1981; Martin GR., 1981). Instead of treating
complex disease processes with a multitude of drugs, each with its own spectrum
of sometimes serious and cumulative side effects, failing components of the
human cytome could be engineered or replaced by stem cells (adult or embryonic)
differentiated into the appropriate cell type.
When the distortion of
cellular metabolism goes beyond a mere dysfunction of a single protein, a
complete replacement of the dysfunctional cells has a better change to restore
the complex and delicate balance and regulation of metabolic processing. The
fine dynamics of spatial and temporal regulation of cellular metabolism and its
response to changing demands of an organism in complex diseases are best met by
replacing the failing part of the cytome with a well balanced cellular
substitute. Those parts of cellular processes which are beyond the reach of
(present-day) drug therapy or which are insufficiently treated by non-cellular
means have the prospect of being restored to a physiologically appropriate
level. With stem cell therapy we would be able to replace a non-functional part
of the human cytome with a set of functioning and dynamically regulated
cellular units.
Several diseases which
currently cannot be treated or cured completely are the target of intensive
research. In diabetes long term insulin replacement therapy does not prevent a
multitude of chronic and severe side effects, such as circulatory
abnormalities, retinopathy, nephropathy, neuropathy and foot ulcers. In
juvenile diabetes however there is an immunological component which complicates
treatment. The prospect to find a cure for diabetes which would restore the
dynamics of insulin production is an important scientific and social challenge
(Heit JJ, 2004).
There is hope for the
development of stem cell therapies in human neurodegenerative disorders (Kim
SU., 2004; Lazic SE, 2004; Lindvall O, 2004). Much research goes into finding a
cure for degenerative diseases such as Parkinson's disease (Drucker-Colin R,
2004; Hermann A, 2004; Roitberg B, 2004). Scientists
are investigating the possibility to treat a failing heart with cellular
cardiomyoplasty (Wold LE, 2004)
When we want to use stem
cells for disease therapy we have to deal with the functional and structural
characteristics of cells which are being used (Baksh D, 2004). The
differentiation of stem cells of either adult or embryonic origin, into mature
and functional cells is a complex and dynamically regulated process.
Understanding the differentiation pathways of embryonic and adult stem cells
and their spatio-temporal dynamics of differentiation
and structural organization will require intensive research (Raff M., 2003).
When using stem cells from an individual which suffers from a degenerative
disease, the disease may not be cured when the same deficient pathway is
activated in the differentiating stem cell. The molecular process may need to
be corrected first in this case, for instance by gene therapy or by using
exogenous stem cells.
Gene therapy also holds
many promises for the therapy of life threatening diseases, but in order to
improve gene
therapy we will need a better understanding on what goes on inside the cell
and what the consequences are on the cellular metabolism when we modify its
function by inserting genes. At this moment monogenic diseases are the target
for gene therapy, but in the future entire parts of pathways may need
reconstruction. The gene is the means to achieve the ultimate goal to change
the cellular metabolism to cure a disease.
The scientific challenges
posed by complex diseases, such as many degenerative and chronic diseases and
cancer will keep scientists busy, far beyond the current scope of present day
science.
Drug
discovery and development
How
to explore and find new directions for research
Conclusion
Figure 3: Exploring the organizational levels of biology.
A linked and overlapping cascade of exploratory systems
each exploring -omes at different organizational levels of biological systems
in the end allows for creating an interconnected knowledge architecture of entire cytomes.
The approach leads to the creation of an Organism Architecture (OA)
in order to capture the multi-level dynamics of an organism.
The future development of
this idea will decide if a Human Cytome Project (HCP) will become reality. The
road from gene to phenotype is not a simple path, but a multidimensional space
built from an extensive web of interacting processes. I can only provide ideas
and explain why it would benefit society and science to explore the cytome in a
more organized and systematic way as is currently being done.. The cellular level of biological organization
deserves more in-depth exploration and quantitative analysis to improve our
understanding of important human disease processes in order to allow us to deal
with the scientific and medical challenges we are facing today and will be
facing in the future.
History of Human Cytome Project set of articles
Previously posted versions, sorted by date
Original
HCP message, 1 December 2003
Update
and first article on website 30 Jan. 2004
Posting
of HCP article version 24 Sept. 2004
Posting
of HCP article version 12 Oct. 2004
Posting
of HCP article version 19 Oct. 2004
Posting
of HCP article version 25 Oct. 2004
Posting
of HCP article version 10 Nov. 2004
Posting
of HCP article version 22 Nov. 2004
Posting
of HCP article version 6 Jan. 2005
Meetings
- Focus
On Microscopy, Philadelphia, USA - 2004
- ISAC
XXII, Montpellier, France - 2004
- European
Microscopy Congress, Antwerp, Belgium - 2004
- EWGCCA, Mol,
Belgium - 2004
- 10th Leipziger
Workshop, Leipzig, Germany - 2005
- 11th Leipziger
Workshop, Leipzig, Germany - 2006
- ISAC
XXIII, Quebec, Canada - 2006
-
Cytomics - Leipziger Workshops
Cytome Research
- Max-Planck-Institut für Biochemie
- Indiana Metabolomics and Cytomics Initiative (METACyt)
- Purdue University
- Bindley Bioscience Center at Discovery Park
- Carnegie Mellon University
- Coriell Institute
- Herzzentrum Leipzig GmbH
- Prince Felipe Centro De Investigation - Cytomics Laboratory
- Catholic Univ. Leuven - Functional cytomics for cell therapy
- Ehime University - Laboratory of Molecular Cell Biology
- Ehime University - Laboratory of Molecular Cell Biology - Section of Cytomics
Additional Information
- Healthcare in Motion
- Biomedical Imaging
- Towards a Human Cytome Project
- Draft: Human Cytome Project
- Innovation and Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products
- New Safe Medicines Faster Project
- Priority Medicines for Europe and the World Project "A Public Health Approach to Innovation"
- Moving Medical Innovations Forward? New Initiatives from HHS
- Cytomics
- E-Cell Project
- Physiome Project
- EuroPhysiome - STEP
- IUPS Physiome Project
- GIOME Project
- Functional genomics
- Cyttron
- National Resource for Cell Analysis and Modeling - NRCAM
- Prediction in Cell-based Systems (Predictive Cytomics)
- Human Genome Project
- Post-Human Genome Project Progress & Resources
- How Many Genes Are in the Human Genome?
- NCBI
- Innovative Medicines Initiative - IMI
- European Science Foundation - ESF
- European Life Sciences Forum - ELSF
Acknowledgments
I am indebted, for their pioneering work on automated digital microscopy and High Content Screening (HCS) (1988-2001 CE), to my former colleagues at Janssen Pharmaceutica (1997-2001 CE), such as Frans Cornelissen, Hugo Geerts, Jan-Mark Geusebroek and Roger Nuyens, Rony Nuydens, Luk Ver Donck, Johan Geysen and their colleagues.
Many thanks also to the pioneers of Nanovid microscopy at Janssen Pharmaceutica, Marc De Brabander, Jan De Mey, Hugo Geerts, Marc Moeremans, Rony Nuydens and their colleagues.
I also want to thank all those scientists who have helped me with general information and articles. My special thanks goes to Andres Kriete, Robert F. Murphy, J. Paul Robinson, Attila Tarnok, and Guenter K. Valet.
References and background
- Van Osta P, Donck KV, Bols L, Geysen J., Cytomics and drug discovery, Cytometry A. 2006 Feb 22;69A(3), pp. 117-118
- Geusebroek, J.-M., Cornelissen, F., Smeulders, A.W. and Geerts, H. (2000), Robust autofocusing in microscopy. Cytometry, 39: 1-9
- I.T. Young and L.J. van Vliet, Recursive implementation of the Gaussian filter, Signal Processing, vol. 44, no. 2, 1995, 139-151
References can be found
here
Some background on the idea can be found
here
Copyright notice and disclaimer
These webpages
represent my personal interests, opinions and ideas, not those of my employer or
anyone else. I have created these web pages without any commercial
goal, but solely out of personal and scientific interest. You may download,
display, print and copy, any material at this website, in unaltered form only,
for your personal use or for non-commercial use within your organization.
Should my web pages or portions of my web pages be used on any Internet or
World Wide Web page or informational presentation, that a link back to
my website (and
where appropriate back to the source document) be established. I expect at
least a short notice by email when you copy my web pages, or part of it for
your own use.
Any information here is provided in good faith but no warranty can be made for
its accuracy. As this is a work in progress, it is still incomplete and even
inaccurate. Although care has been taken in preparing the information contained
in my web pages, I do not and cannot guarantee the accuracy thereof. Anyone
using the information does so at their own risk and
shall be deemed to indemnify me from any and all injury or damage arising from
such use.
To the best of my knowledge, all graphics, text and other presentations not
created by me on my web pages are in the public domain and freely available
from various sources on the Internet or elsewhere and/or kindly provided by the
owner.
If you notice something incorrect or have any questions, send me an email.
By Peter Van Osta
Email: pvosta at gmail dot com
A first draft was published
on Monday, 1 December 2003 in the bionet.cellbiol
newsgroup. I posted regular updates of this text to the bionet.cellbiol
newsgroup.
Latest revision on 26 May 2018